
Graphic Abstract
Introduction
A virus must survive in the environment outside its host to spread. Its persistence, and thus the extent and speed of transmission, depends on three key factors: viral characteristics, the nature of the surface it contaminates, and the environmental conditions (1). Therefore, the widespread transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may be facilitated by its ability to persist on surfaces. This persistence can last from hours to days, particularly for variants such as Omicron, as reported in studies (2–5). SARS-CoV-2 viral proteins, shed by infected individuals through fecal excretion into wastewater, are widely detectable in hospital-adjacent aerosols, soils, and on 34.1 % of isolation-ward surfaces (6–9).
At present, the detection of SARS-CoV-2 viral proteins in environmental samples relies primarily on mass spectrometry (MS), electrochemical immunosensors, and reverse transcription quantitative polymerase chain reaction (RT-qPCR) for confirmatory analysis (17,18). Owing to its high sensitivity and specificity, MS has become an important tool for identifying viral proteins in wastewater (17). Studies have shown that MS can detect signature peptides of virus-specific proteins—such as the non-structural protein NSP3—5–6 days before clinical case reports, offering an early-warning capacity (17). Nevertheless, environmental protein concentrations are extremely low and are easily affected by organic matter, suspended particles, and pH fluctuations; therefore, MS requires rigorous sample pre-treatment (e.g., concentration, enzymatic digestion, decontamination) to improve recovery (19). Parallel reaction monitoring (PRM) has been developed as a targeted quantification method, and its scan mode is well-suited for analyzing low-intensity peptides. With the addition of stable isotope-labeled internal peptides, PRM can detect targeted peptides in a manner similar to multiple reaction monitoring (MRM) (27).
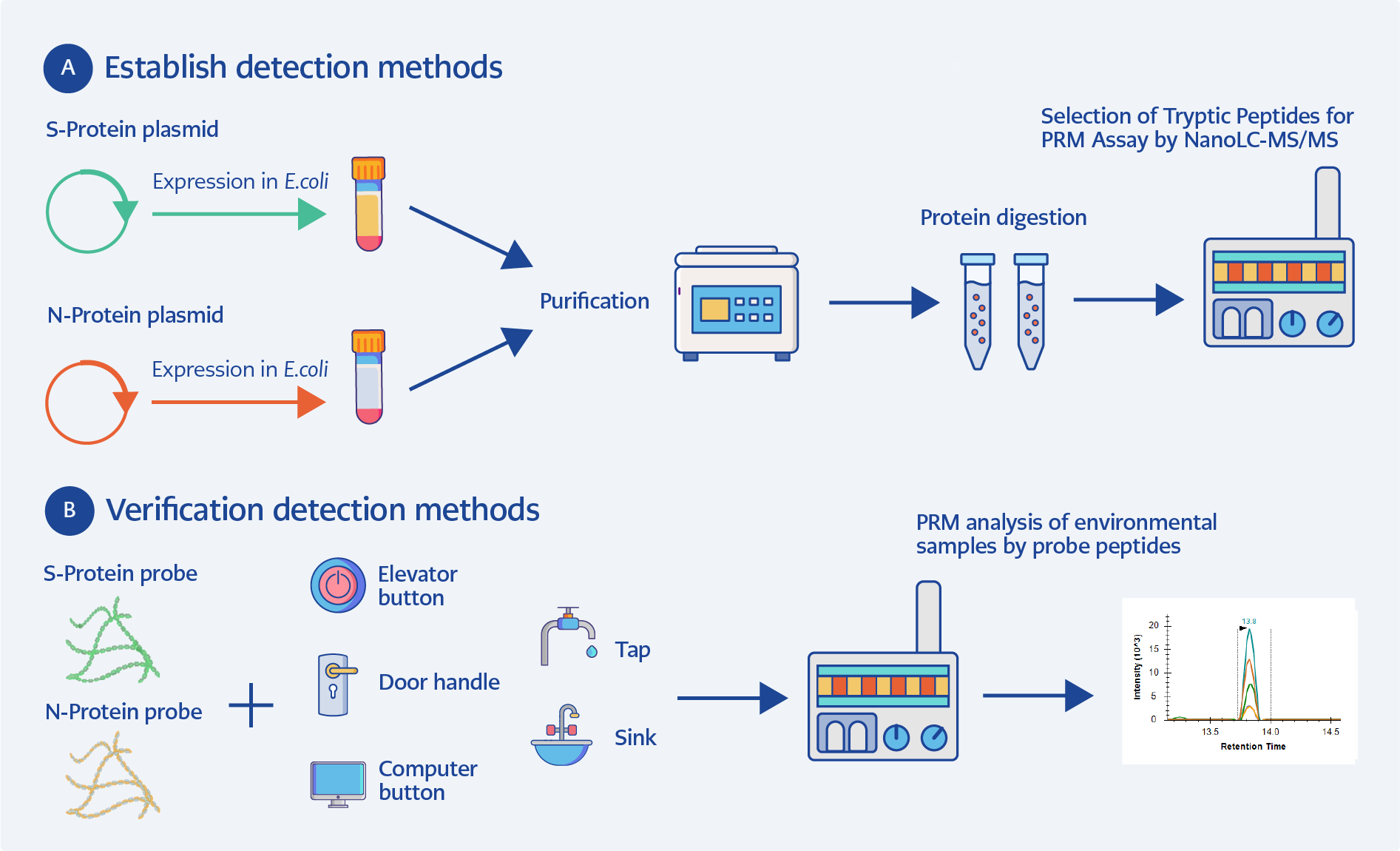
Figure 1. SARS-CoV-2 peptide probe-based PRM detection workflow. (A) Detection method development; (B) Detection method validation.
This study developed a sensitive and targeted nano–liquid chromatography–tandem mass spectrometry (nanoLC-MS/MS) parallel reaction monitoring (PRM) workflow for the detection of SARS-CoV-2 proteins in environmental samples. Specific peptide sequences derived from the spike 1 (S1) and nucleocapsid (N) proteins were evaluated as candidate targets for detection. The performance of the developed method was subsequently evaluated for the trace detection of residual SARS-CoV-2 proteins in various environmental samples (Figure 1).
Materials and Methods
Bacterial Strains and Growth Conditions
Escherichia coli cells were grown at 37°C in Luria-Bertani (LB) broth (Difco™, BD Biosciences, Sparks, MD, USA; Cat No. 214906). When necessary, antibiotics were added to bacterial cultures at a final concentration of 50 µg/mL of kanamycin (Km) (Solarbio Life Sciences, Beijing, China; Cat No. K8020). Escherichia coli DH5α (TransGen Biotech Co., Ltd., Beijing, China; Cat No. CD201-01) and BL21-Gold (DE3) (XY Bioscience Co., Ltd., Beijing, China; Cat No. XY1028) were used as hosts for plasmid DNA preparation and protein production, respectively.
Isolation of SARS-CoV-2-S1-His6 and SARS-CoV-2-N-His6 Proteins
Escherichia coli BL21-Gold (DE3) harboring the plasmids pET28a-SARS-CoV-2-S1 (spike 1) (pDY102) and pET28a-SARS-CoV-2-N (pDY104) was grown at 37°C with shaking in 500 mL LB medium until an optical density at 600 nm (OD600) of 0.5 was reached. Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Solarbio, Beijing, China; Cat No. I8070) was then added to a final concentration of 0.5 mM, and the culture was incubated for 2 h.
Cells were harvested, washed once with phosphate-buffered saline (PBS) (Solarbio, Beijing, China; Cat No. P1020), and resuspended in 10 mL PBS. Cell lysis was performed by sonication. The whole-cell lysate was used for purification of SARS-CoV-2-S1-His6 and SARS-CoV-2-N-His6 proteins using a His-Select Ni–nitrilotriacetic acid (Ni–NTA) Sepharose column (BBI Solutions, Crisp Research Centre, Sittingbourne, UK; Cat No. C600792) according to the manufacturer’s instructions.
Briefly, proteins were eluted with elution buffer containing 50 mM NaH2PO4 (Solarbio, Beijing, China; Cat No. S5830), pH 8.0, 300 mM NaCl (Solarbio, Beijing, China; Cat No. S8212), and 100 mM imidazole (Solarbio, Beijing, China; Cat No. I8090). Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis was performed in 10% separating gel to verify the molecular weight.
Purified SARS-CoV-2-S1-His6 and SARS-CoV-2-N-His6 proteins were stained with Coomassie Brilliant Blue (CBB) R-250 (Solarbio, Beijing, China; Cat No. C8430) and compared with a standard pre-stained protein ladder according to the method described by Laemmli (28).
In-Solution Targeted Protein Digestion
The targeted protein was suspended in 0.1% (w/v) RapiGest SF (Waters, Milford, MA, USA) solution containing 50 mM ammonium bicarbonate (NH4HCO3) (Solarbio, Beijing, China; Cat No. A0240) and reduced in 5 mM 1,4-Dithiothreitol (DTT) (Solarbio, Beijing, China; Cat No. D8220) at 56°C for 30 min. After cooling, alkylation was carried out by incubating the sample in the dark with 10 mM iodoacetamide (IAA) (Solarbio, Beijing, China; Cat No. I8010) for 30 min.
An additional 5 mM DTT was then added and incubated for 15 min at room temperature. Labeled peptides were added to the solution at a ratio of 4:1 relative to unlabeled peptides. Sequencing-grade modified trypsin was subsequently added at a ratio of 1:50 (trypsin:protein), and digestion was performed overnight at 37°C.
After digestion, the solution was acidified with trifluoroacetic acid (Aladdin, Shanghai, China; Cat No. U320373) and incubated at 37°C for 1 h to degrade RapiGest SF. Following centrifugation at 13,000 g for 10 min, the solution was transferred to another tube and dried under vacuum.
Collection and Preparation of the Environmental Sample
Samples were collected from five sites: elevator buttons, door handles, computer mouse buttons, taps, and sinks. The test surfaces were wiped with a wetted sterile polyester swab, which was then immersed in 1 mL PBS.
A 500 μL aliquot of the environmental sample solution was supplemented with NH4HCO3 and DTT to final concentrations of 50 mM and 5 mM, respectively, and heated at 95°C for 5 min. After cooling, alkylation was performed by incubating the sample in the dark with 10 mM IAA for 30 min. An additional 5 mM DTT was then added and incubated for 15 min at room temperature.
Sequencing-grade modified trypsin was added to a final concentration of 0.01 μg/μL, and digestion was performed overnight at 37°C. After digestion, the solution was acidified with trifluoroacetic acid and dried under vacuum.
The dried residue was dissolved in 250 μL of H2O, 0.1% formic acid (FA) (Thermo Fisher Scientific, Waltham, MA, USA; Cat No. 036504-AP) and mixed with an equal volume of the targeted viral peptides.
NanoLC−MS/MS Analyses
A Q Exactive ultra-high-field (HF) mass spectrometer coupled with an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific, Waltham, MA, USA) was used for data-dependent acquisition (DDA) experiments. Each sample was loaded onto a trap column (100 μm × 2 cm, packed with ReproSil-Pur C18-AQ, 3 μm) using mobile phase A (0.1% FA in water).
Peptide separation was performed on an analytical C18 column (75 μm × 12 cm, packed with ReproSil-Pur C18-AQ, 3 μm) at a flow rate of 300 nL/min with the following gradient: 4% to 40% mobile phase B (0.1% FA in 80% acetonitrile; Thermo Fisher Scientific, Waltham, MA, USA; Cat No. 047138-M1) over 9 min, 40% to 90% B over 5 min, and holding at 90% B for 5 min.
The mass spectrometer was operated in positive-ion mode. Full MS scans were acquired at a resolution of 30,000, with an automatic gain control (AGC) target of 3 × 106 and a maximum injection time of 100 ms. The top 20 most intense ions were selected for MS/MS analysis.
MS/MS spectra were generated using higher-energy collisional dissociation (HCD) with a normalized collision energy of 27%. Fragment ion scans were collected at a resolution of 15,000, with an AGC target of 1 × 105, a maximum injection time of 100 ms, and an isolation window of 1.6 m/z.
Raw DDA data were processed using Proteome Discoverer (PD) software version 2.2 (Thermo Fisher Scientific, Waltham, MA, USA). Trypsin was specified as the enzyme with up to two missed cleavages allowed. The precursor mass tolerance was set to 10 ppm and the fragment mass tolerance to 0.02 Da. Carbamidomethylation of cysteine was set as a fixed modification, and oxidation of methionine as a variable modification. Results were filtered using the percolator node to achieve a false discovery rate (FDR) of <1%.
Targeted Analysis by Parallel Reaction Monitoring (PRM)
Parallel reaction monitoring analysis was performed using a Q-Exactive HF mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with an UltiMate 3000 LC system. Each sample was loaded onto a trap column (100 μm × 2 cm, packed with ReproSil-Pur C18-AQ, 3 μm) with phase A (0.1% FA in water).
Peptide separation was carried out on a C18 column (75 μm × 12 cm, packed with ReproSil-Pur C18-AQ, 3 μm) at a flow rate of 300 nL/min with the gradient of 4-40% phase B (0.1% FA in acetonitrile) for 9 min, 40 to 90% phase B for 5 min, and 90% phase B for 5 min.
Full scans were acquired at a resolution of 30,000 with an AGC target of 3 × 106 and a maximum injection time of 100 ms. PRM scans were acquired at a resolution of 30,000 with an isolation width of 1.0 m/z, an AGC target of 5 × 105, a maximum injection time of 500 ms, and a normalized collision energy of 27% in an HCD cell. The inclusion list is provided below.
Parallel reaction monitoring data were analyzed using Skyline daily (64 bit) version 19.1.9.350. A spectral library of reference MS/MS spectra was generated from the DDA dataset. Peaks were manually inspected based on retention time, transitions, and MS/MS spectra. At least five transitions were considered for each peptide.
Evaluation of Limit of Detection
The optimized PRM method was applied using mock virus and peptide standards to evaluate the limit of detection for viral contamination in various environmental samples. Peptide standards containing two 13C15N-labeled residues were spiked into environmental samples.
Quality criteria for identification of targeted peptides included the retention-time difference between labeled and unlabeled peptides and the dot-product score (dopt) in Skyline. The retention-time difference was required to be ≤0.1 min. The dopt value provided a correlation score between the measured product-ion peak areas and those in the library spectra.
In this study, a dopt value ≥0.9 was considered a positive identification. These criteria ensured analytical accuracy and minimized false-positive results. Mock virus samples were spiked post-digestion, and peptide responses were evaluated from 0.1 fmol to 100 fmol on-column. Samples were analyzed in triplicate.
Each run, including column equilibrium, sample injection, and peptide separation, required 30 min, with a 20 min acquisition window, enabling both rapid detection and subsequent quantification.
Data analysis
Parallel reaction monitoring results were imported into Skyline (version 4.1) for targeted extracted ion chromatogram (XIC) analysis. Five fragment-ion transitions were selected for each target peptide based on fragment-ion intensity, signal-to-noise ratio, and co-elution of native and heavy-labeled peptides.
Using the optimized PRM method, one peptide sequence per protein was used for absolute quantification, and a second peptide was used to confirm identity and specificity.
Results
Isolation of SARS-CoV-2-S1 and SARS-CoV-2-N from E. coli
For the expression of mature SARS-CoV-2-S1 and SARS-CoV-2-N genes in E. coli, pET28a-SARS-CoV-2-S1 (pDY102) and pET28a-SARS-CoV-2-N (pDY104) were introduced into E. coli BL21(DE3). Expression of SARS-CoV-2-S1 (~75 kDa) and SARS-CoV-2-N (~50 kDa) gene was analyzed following induction with 0.5 mM IPTG.
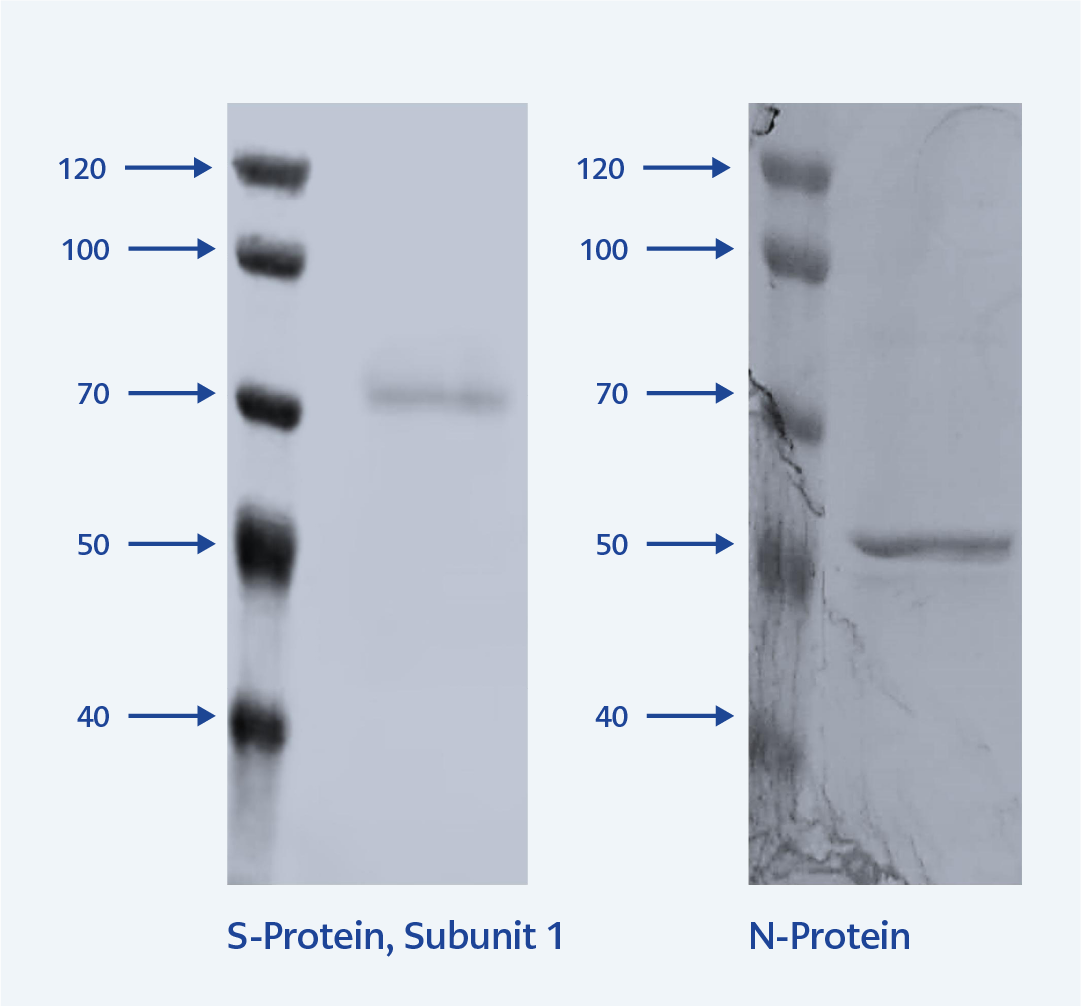
Figure 2. SDS-PAGE analysis of purified SARS-CoV-2 S1 and N proteins.
M: Molecular weight marker, S1: Purified spike protein subunit 1 (left), and NP: Purified nucleocapsid protein (right).
The SARS-CoV-2-S1 protein and SARS-CoV-2-N proteins were purified using a His-Select Ni-NTA-Sepharose column and visualized by SDS-PAGE gel (Figure 2).
Selection of Tryptic Peptides for PRM Assay
A standard-driven strategy was employed to identify candidate sequences to serve as surrogate peptides for targeted nanoLC-MS/MS PRM analysis (29). Ideally, target peptides (usually 5−25 amino acids) should be unique to SARS-CoV-2 proteins and derived from different regions of the protein of interest.
To select optimal candidate peptides for PRM quantification, recombinant SARS-CoV-2 S1 protein and SARS-CoV-2-N protein were digested with trypsin. The resulting peptides were analyzed by nanoLC-MS/MS using DDA on an Orbitrap instrument (Q Exactive HF-X Hybrid). For the S1 protein, peptides covering 78.04% of the sequence were detected, whereas for the N protein, tryptic digestion resulted in 64.99% sequence coverage.
Based on this extensive sequence coverage and peptide response, a list of tryptic peptide targets was generated for subsequent PRM analysis. The identified peptides were further analyzed using NCBI BLAST to confirm their specificity for SARS-CoV-2.
Heavy-labeled peptides were synthesized according to the targeted sequences of S1 and N proteins. The C-terminal lysine and arginine residues were replaced by d8-lysine and d10-arginine, respectively. Because the labeled and unlabeled peptides have nearly identical chemical properties, they exhibited highly consistent chromatographic behavior in reversed-phase LC systems. Excess labeled peptides were added to provide a stable internal standard for detecting the corresponding unlabeled peptides.

Figure 3. Sequence coverage used for target peptide selection in PRM assay development for SARS-CoV-2 S1 and N proteins.
The amino acid sequences of the SARS‑CoV‑2 S1 protein (top) and nucleocapsid (N) protein (bottom) are shown. Peptides identified through tryptic digestion and data-dependent acquisition (DDA) are mapped to the respective protein sequences. Selected target peptides for the final PRM assay are highlighted with a brown background: FASVYAWNR and QIAPGQTGK from the S1 protein, and NPANNAAIVLQLPQGTTLPK and GPEQTQGNFGDQELIR from the N protein. These peptides were chosen based on criteria including high MS signal intensity, specificity for SARS-CoV-2, and the absence of missed tryptic cleavage sites.
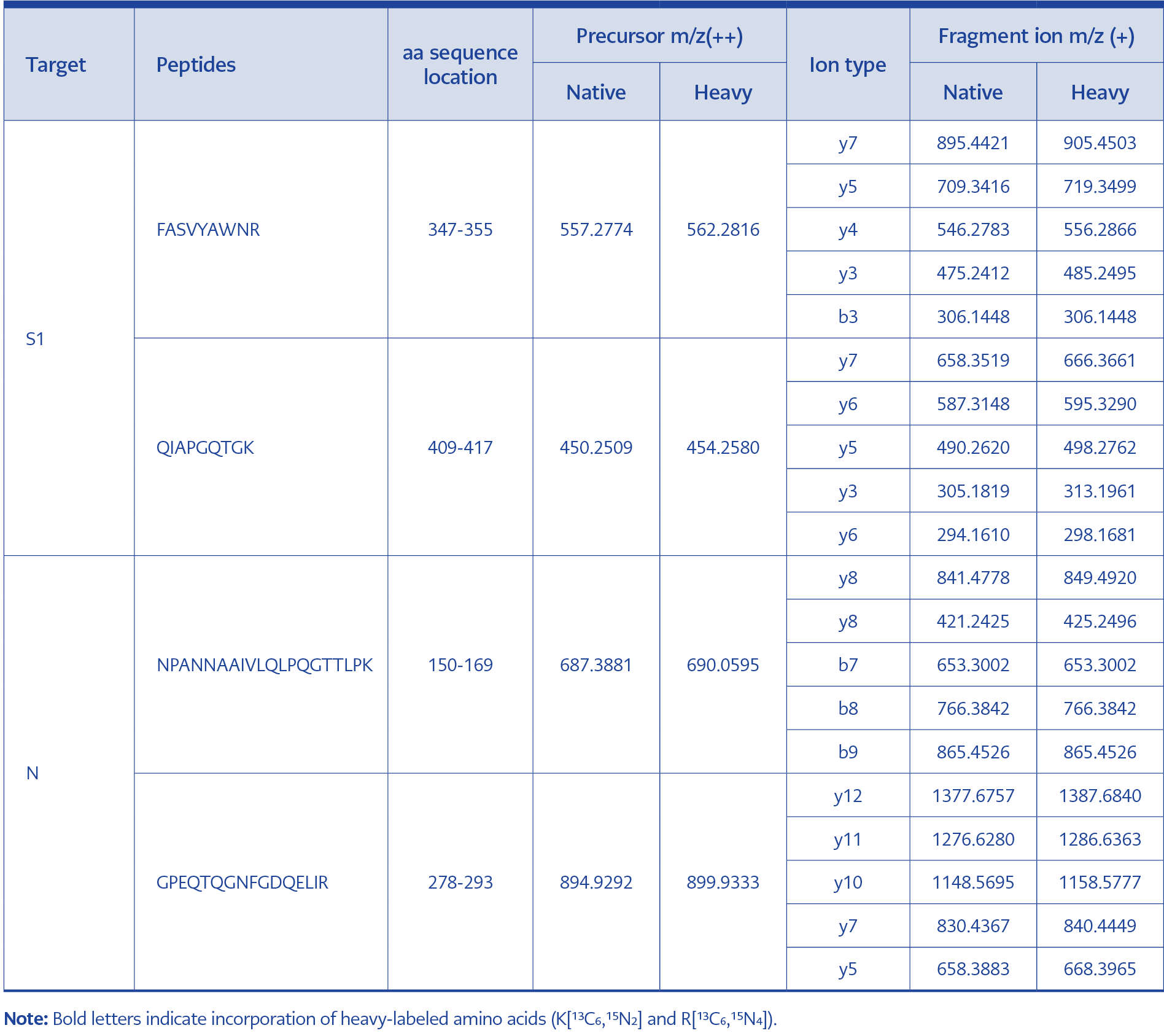
Table 1. Fragment ion transitions used for nanoLC-MS/MS (PRM) analysis of S1 and N peptides.
Based on the following criteria—1) High intensity, 2) Specificity, 3) No modification, 4) No missed cleavage sites—the peptides FASVYAWNR and QIAPGQTGK from the S1 protein and NPANNAAIVLQLPQGTTLPK and GPEQTQGNFGDQELIR from the N protein were selected for PRM analysis (Figure 3, Table 1).
Detection of Mock Virus in Environmental Samples
Applying the optimized PRM method, heavy peptide standards were used to evaluate the limit of detection for mock virus contamination in environmental samples. The nanoLC-PRM method was optimized to measure viral peptides in six different environmental matrices.
Mock virus peptidomes were mixed with background matrices to final concentrations of 0.1 fmol/μL, 1 fmol/μL, 10 fmol/μL, and 100 fmol/μL. A total of 5 μL from each of the six sets of samples (18 samples in total) was injected into the nanoLC-MS/MS system using the PRM method.
For the S1 protein, FASVYAWNR and QIAPGQTGK were used for confirmation. FASVYAWNR was detected at the lowest concentration of 1 fmol/μL in one of the six environmental samples, whereas QIAPGQTGK was detected at a concentration of 1 fmol/μL in all six environmental samples.
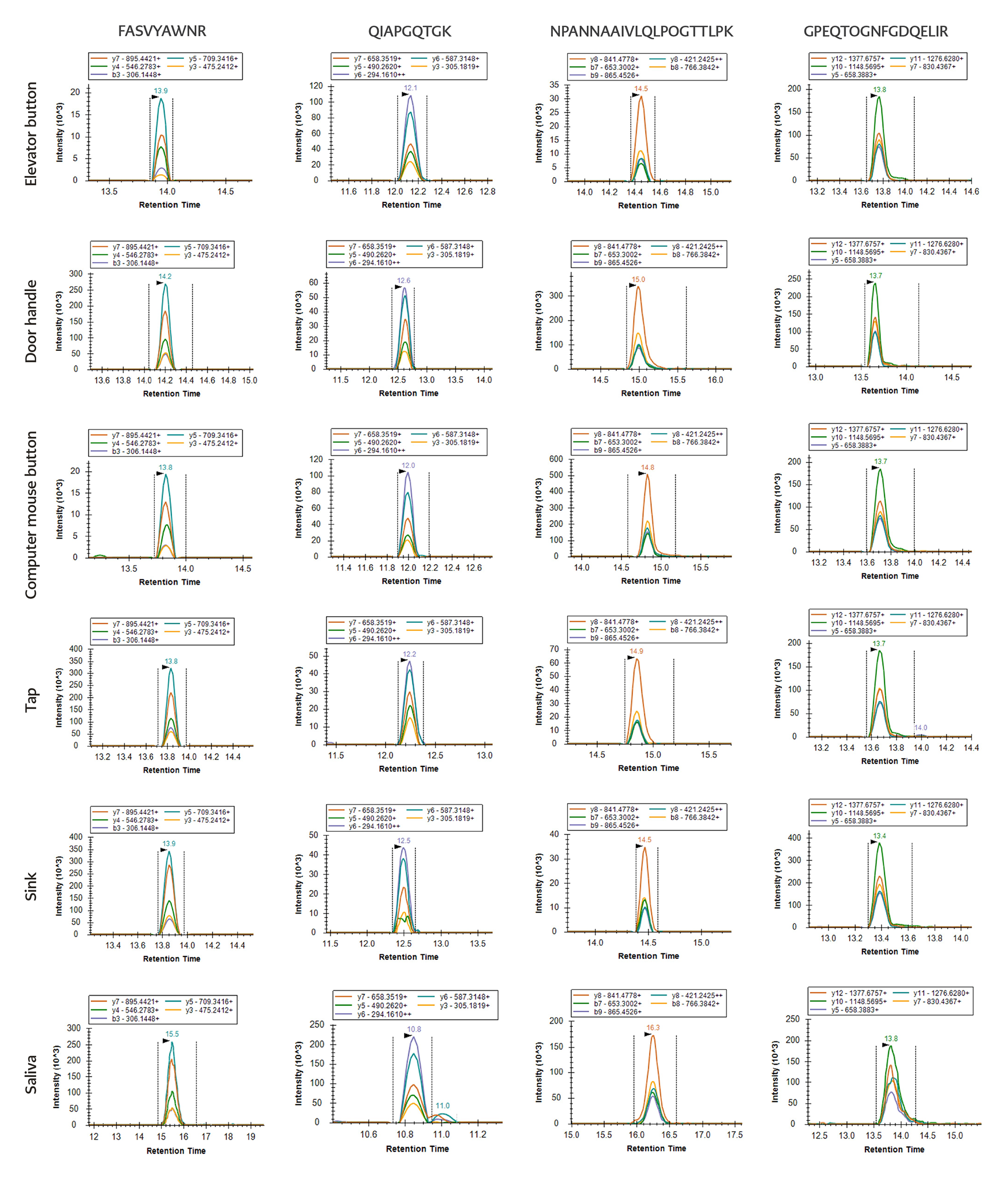
Figure 4. PRM analysis by four peptides selected for SARS CoV-2 detection in environmental samples.
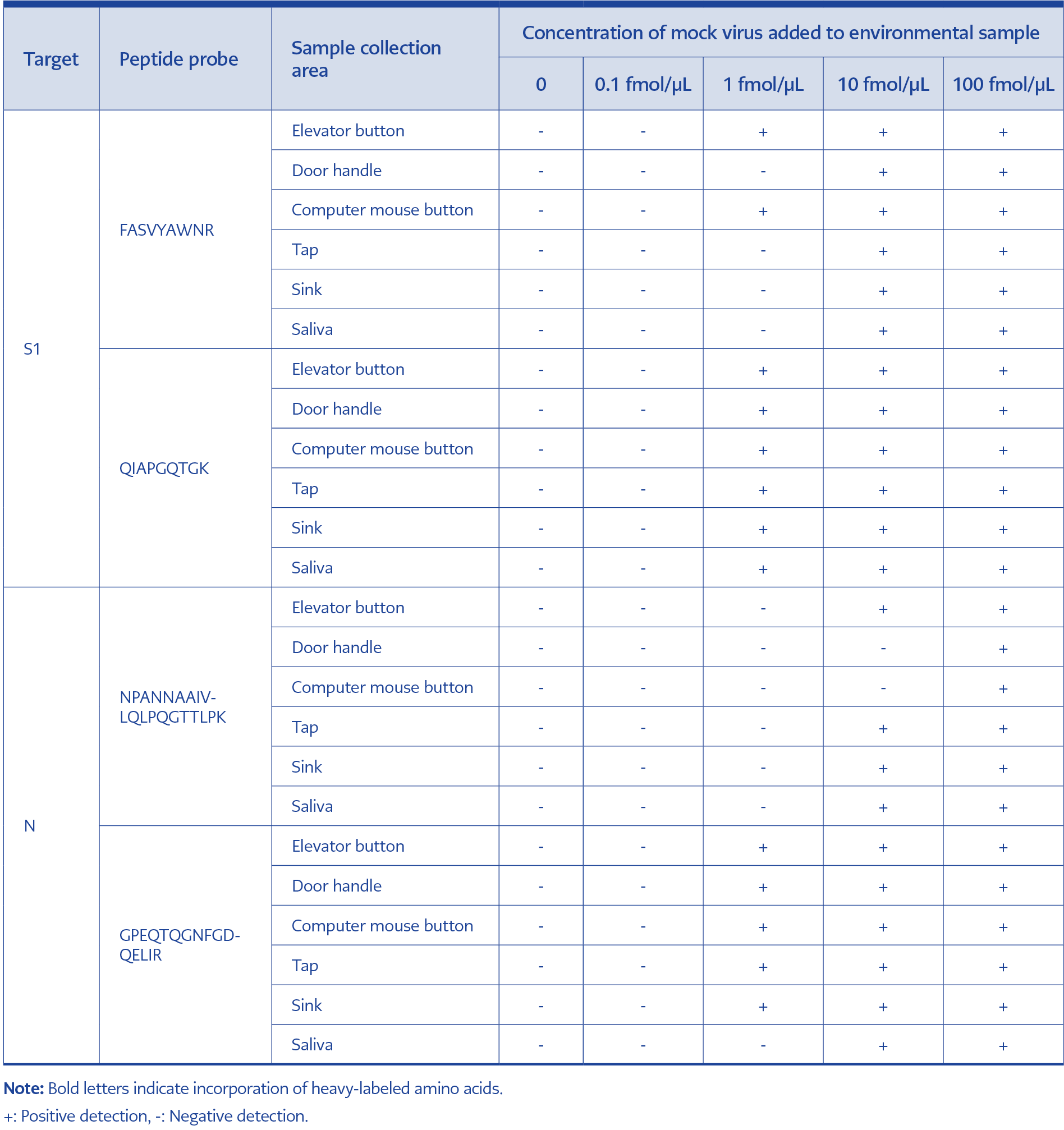
Table 2. Detection results for SARS-CoV-2 in environmental samples using optimized S1 and N peptide standards.
For the N protein, NPANNAAIVLQLPQGTTLPK and GPEQTQGNFGDQELIR were used for confirmation. NPANNAAIVLQLPQGTTLPK was detected at a lowest concentration of 10 fmol/μL in four of the six environmental samples, while GPEQTQGNFGDQELIR was detected at the same concentration in five of the six environmental samples (Figure 4, Table 2).
Based on these results, QIAPGQTGK and GPEQTQGNFGDQELIR were selected as the primary detection peptides because of their higher sensitivity.
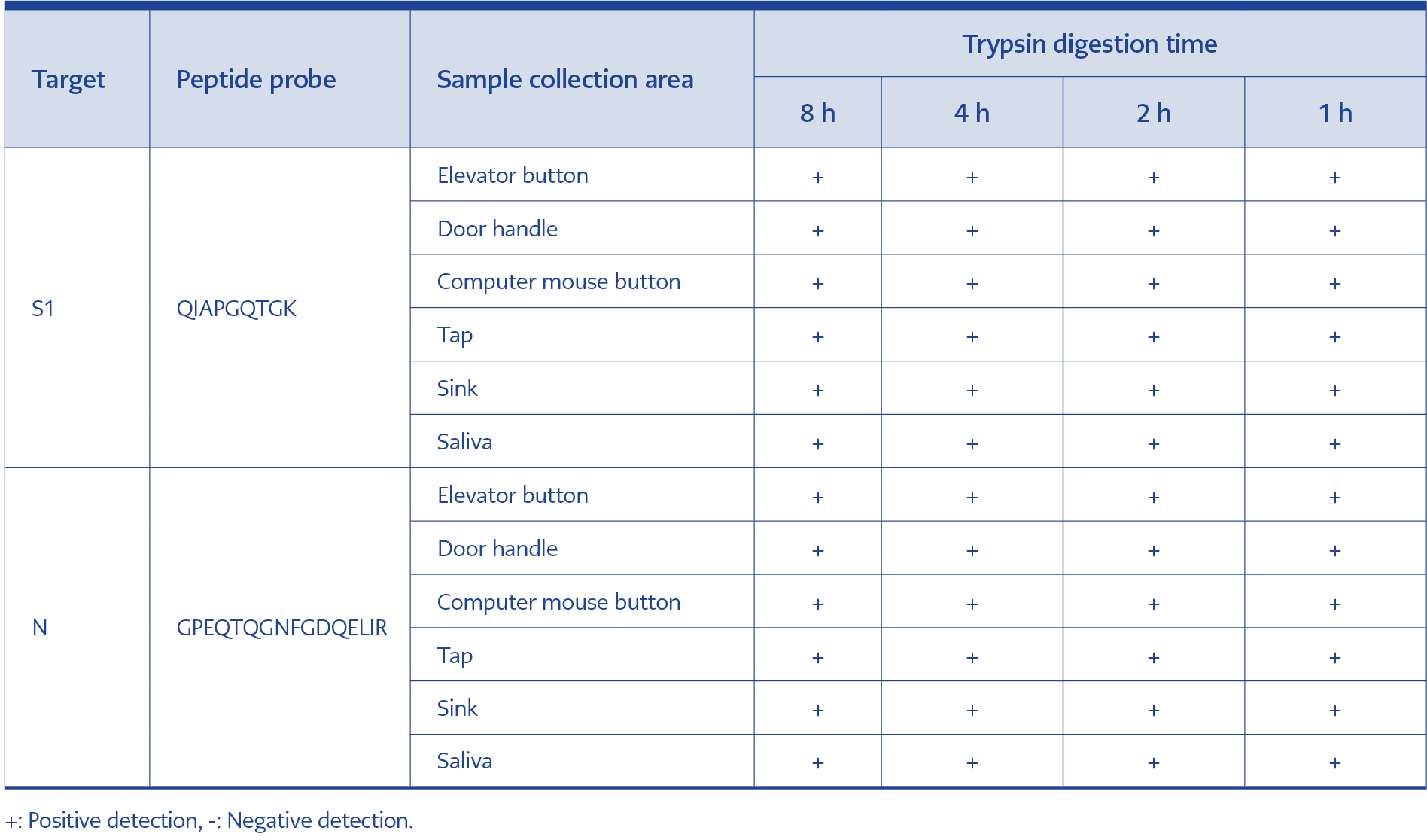
Table 3. Time-course analysis of trypsin digestion and detection of SARS-CoV-2 at the 10 fmol/μL.
To evaluate the effect of digestion time on detection sensitivity, samples were digested with trypsin for 8 h, 4 h, 2 h, or 1 h. Peptides QIAPGQTGK and GPEQTQGNFGDQELIR were used to detect SARS-CoV-2 based on the previously established dataset. A concentration of 1 fmol/μL could not be detected in all environmental samples when digestion times were shorter than 8 h, whereas the 10 fmol/μL level was detectable at all digestion times ranging from 1 to 8 h (Table 3).
Discussion
Detection of viral proteins in environmental samples is important for understanding viral transmission dynamics, elucidating infection-associated pathogenic mechanisms, and assessing potential public health risks. Although most studies on viral proteins and their pathogenic roles have focused on in vivo or cell-culture systems, accumulating evidence suggests that viral proteins can retain biological activity in extracellular environments after being released from infected cells. These extracellular viral proteins may influence viral dissemination and host immune modulation. Environmental protein detection strategies may therefore enhance surveillance by identifying subclinical viral shedding, revealing extracellular virulence mechanisms, and enabling targeted interventions to disrupt both direct transmission and environmental persistence.
Currently, the standard diagnostic approach relies on amplification and detection of viral RNA using reverse transcription-polymerase chain reaction (RT-PCR). At the same time, numerous rapid, accurate detection methods have been developed. For example, nucleic acid-based assays such as ID NOW (10) have been reported to exhibit higher sensitivity than RT-PCR in some settings, and reverse transcription recombinase polymerase amplification (RT-RPA) has been shown to detect even a single copy of viral RNA (11).
Protein-based detection strategies have also been explored. Field-effect transistor (FET) biosensors have demonstrated detection limits of 102 copies/mL of virus (12), and terahertz plasmonic metasensor systems can detect viral proteins at the femtomolar level (13).
Antigen-detection assays and RT-PCR are most commonly used for virus detection during the acute phase of infection. In the case of SARS-CoV-2, structural proteins such as S, N, M, and E—and their corresponding—have been widely used as diagnostic targets. In addition, complementary methods have been developed for SARS-CoV-2 detection, including reverse transcription loop-mediated isothermal amplification (RT-LAMP), electrochemical and optical biosensors for RNA detection, and assays targeting whole viruses or viral proteins (31,32). Antigen tests have helped alleviate testing bottlenecks in countries such as Singapore, South Korea, and China.
Liquid chromatography–mass spectrometry (LC-MS) is well-suited for the analysis of large, polar, ionic, thermally unstable, and involatile compounds, including antigenic proteins. Consequently, mass spectrometry-based systems offer an attractive alternative for viral protein detection. They have previously been investigated for the identification of respiratory viruses and represent a promising strategy for rapid and sensitive detection of SARS-CoV-2.
Our results indicate that femtomolar-level sensitivity can be achieved for SARS-CoV-2 proteins in complex matrices using the PRM technique. To determine whether this sensitivity is sufficient for environmental detection, we considered the viral titers used to prepare mock samples. The entire RT-PCR process typically takes 1–2 h, including 20–40 min for RNA extraction and 30–90 min for PCR amplification. Although highly sensitive, RT-PCR-based environmental monitoring primarily detects residual viral genetic material and cannot reliably distinguish infectious viral particles from fragmented RNA.
In contrast, structural proteins such as the SARS-CoV-2 S and N proteins are integral components of intact virions. Detection of specific viral peptides derived from these proteins may therefore provide stronger evidence of the presence of intact viral particles in environmental samples. Protein-level detection may thus help identify potential transmission sources and support targeted public-health interventions, such as localized disinfection strategies.
In the workflow developed in this study, the entire procedure—including protein digestion (1 h) and detection (30 min)—can be completed within 2 h (Table 3). Therefore, this method is comparable to RT-PCR in terms of time, cost, and sample throughput.
Selection of representative peptides is a critical step in targeted proteomics. The SARS-CoV-2 S and N proteins represent important targets for virus detection. The binding of the S1 subunit to a host ACE2 receptor can destabilize the prefusion trimer, leading to shedding the S1 subunit, and transition of the S2 subunit to a highly stable post-fusion conformation (33). To engage host receptors, the receptor-binding domain (RBD) of the S1 subunit undergoes hinge-like conformational movements that transiently expose or conceal receptor-binding determinants (33,34).
The SARS-CoV-2 N protein is highly conserved among viral variants and plays an essential role in viral RNA replication and packaging into new virions (35). Several specific peptides were identified from tryptic digestion of the S1 and N proteins (Table S1) and can serve as candidate targets for detection of different samples.
Nanoflow chromatography is frequently used in proteomics workflows to achieve the highest sensitivity. High-quality chromatographic separation is critical because peak shape and resolution influence the ability of the MS system to sample distinct peptides. Column length, gradient length, and peptide characteristics all affect chromatographic performance. Although longer columns and gradients generally increase peptide identification, the degree of improvement becomes dependent on both parameters and is diminished at longer columns and gradients (36). In the present study, chromatographic performance was optimized for the selected column and gradient conditions, and peptide characteristics such as binding strength were considered during probe selection (Table 2).
PRM methods require less prior knowledge than many other targeted proteomic approaches because only precursor masses need to be defined to create an MS instrument method. When multiple precursor ions are screened for a given protein, PRM data can subsequently be matched against protein databases to confirm protein identification. This approach reduces the initial optimization effort compared with other targeted workflows (37). Consequently, PRM provides a rapid and flexible strategy for developing detection methods for SARS-CoV-2 or other emerging viruses.
Previous studies have estimated that each SARS-CoV-2 virion carries approximately 120 copies of the S protein, corresponding to ~40 trimeric spike complexes on the viral surface (since each trimer contains three S-protein monomers) (38). In addition, each virion contains a helical ribonucleoprotein (RNP) complex packaging the ~30 kb viral RNA genome. This RNP structure is composed of roughly 38 globular N protein-RNA complexes, each containing approximately 12 N-protein monomers, resulting in an estimated total of approximately 450–460 copies of N protein per virion (39). Based on these estimates, the limit of detection achieved in this study (1 fmol/µL range on-column) corresponds to 2–8 × 10-15 virus/L.
From an economic perspective, although the unit cost of nanoLC-MS/MS PRM (approximately USD 30–50 per sample) is higher than that of RT-PCR (USD 5–10) and rapid antigen assays (USD 1–3), its attomolar-level sensitivity (1 fmol, ≈ 2–8 × 10-15 virion L-1) enables detection of SARS-CoV-2 in environmental matrices at the incipient stage of contamination. When considered as an early-warning surveillance strategy rather than a routine diagnostic test, high-sensitivity PRM monitoring at critical locations could offer a favorable cost-benefit ratio (>1:500) by minimizing downstream lockdown expenditures and preserving economic continuity.
In this study, S1 and N proteins were expressed and purified using an E. coli expression system, and specific peptide candidates were screened using DDA analysis. Two representative peptides were selected for each protein and synthesized with stable-isotope labels to serve as internal standards. These labeled peptides were then used for targeted detection of viral peptides in environmental samples using nanoLC-MS/MS PRM. The method enabled detection of SARS-CoV-2 peptides at the fmol/μL level within 2 h. Using the peptides QIAPGQTGK and GPEQTQGNFGDQELIR as quantitative standards, we established a targeted nanoLC-MS/MS PRM workflow for rapid detection of SARS-CoV-2 in environmental samples. These results support the further development of this approach as an in vitro method for detecting residual SARS-CoV-2 contamination on environmental surfaces.