Introduction
Hepatitis B virus (HBV) infection is the leading cause of chronic liver disease worldwide. HBV-infected patients are at risk of developing liver cirrhosis and hepatocellular carcinoma (HCC) for life. Today, pegylated interferon (Peg-IFN) and nucleos(t)ide analogues (NAs) are used in the treatment of patients with chronic hepatitis B (CHB). Both treatment options have limitations. Nucleo(t)id analogs group antivirals used in treatment, especially tenofovir disoproxil fumarate (TDF), entecavir (ETV) and tenofovir alafenamide fumarate (TAF), effectively suppress HBV DNA levels and have been shown to prevent the progression of the disease to cirrhosis, liver fibrosis, and cirrhosis. They cannot be eliminated. Nucleo(t)id analogs exert effective viral suppression but have little effect on covalently closed circular DNA (cccDNA), the stable episomal form of the HBV genome in hepatocytes. Therefore, the cure rate with NAs is low (3-5%), and long-term treatment is required. Peg-IFN therapy, on the other hand, is for a limited time and is effective at different stages of the HBV life cycle. However, it has a low response rate, and the drug is difficult to tolerate (1-4). Our goal with treatment in patients with chronic HBV infection is to reach the functional cure defined as continuous loss of HBsAg. Complete cure is defined as permanent loss of HBsAg and elimination of undetectable serum HBV DNA and cccDNA. Although measuring intrahepatic cccDNA activity by liver biopsy is necessary to demonstrate this condition, serum biomarkers reflecting cccDNA levels can be used instead. As candidates, HBV RNA, hepatitis B core-associated antigen (HBcrAg), and quantitative HBsAg are under investigation (2-4). The inability to reach a functional cure with current treatment options necessitates the development of new antivirals. New antiviral treatments can be grouped under two headings: direct-acting antiviral drugs and immunotherapeutic approaches.
HBV Life Cycle
Viral entry occurs via binding of the pre-S1 domain of L-HBsAg to the sodium taurocholate co-transporting polypeptide (NTCP) receptor on the hepatocyte plasma membrane, after which the viral particles are separated from its envelope, and the nucleocapsids are transported to the nucleus of infected cells. They release partially double-stranded loose circular DNA. Host DNA packages cccDNA into chromatin by histone and non-histone proteins to form a viral minichromosome. This minichromosome is the HBV transcription template and plays a key role in the HBV life cycle. The pgRNA is encapsulated by polymerase and reverse transcribed into viral negative stranded DNA. The positive-stranded DNA is then synthesized to form a partially double-stranded loose circular DNA. Following viral DNA synthesis, mature nucleocapsids are either recycled to the nucleus to preserve the cccDNA pool or packaged with envelope proteins and exported as infectious virions (5-9).
Direct Acting Antivirals
HBV Entry Inhibitors
HBV entry into hepatocytes is the first step of HBV infection. NTCP, the functional receptor of HBV, interacts specifically with the preS1 region of the HBV envelope protein and allows HBV to be taken into hepatocytes (4). Bulevirtide (BLV) is a synthetic N-acetylated lipopeptide containing the NTCP-binding pre-S1 domain of the large HBsAg envelope protein. Bulevirtide competitively binds to NTCP and blocks de novo HBV infection by competing with the pre-S1 motif for binding with NTCP. Bulevirtide 2 mg/day treatment was approved for the treatment of chronic delta hepatitis in the European Union in 2020 (1,10). In the multicenter phase 2 MYR202 study, 120 patients with chronic delta hepatitis treated with TDF were randomized to different doses of BLV (2, 5, or 10 mg/day) for 24 weeks, followed by either TDF therapy or TDF monotherapy. At the end of the study, HBsAg loss was not achieved in both treatment arms (10,11). In the phase 2 study MYR203, 48 weeks of treatment with different doses of BLV with or without Peg-IFN α was given. Patients with chronic HBV/HDV coinfection were grouped as Peg-IFN a monotherapy or BLV monotherapy + BLV’s TDF or Peg-IFN combinations, and HBsAg loss was observed only in patients who were given Peg-IFN a combined BLV (12).
In another phase 2 MYR204 study, a decrease of more than 1 log IU/ml from baseline in HBsAg levels in patients given Peg-IFN α monotherapy or combined BLV was higher in the Peg-IFN α and BLV combined arm (13). In the MYR301 Phase 3 study, patients with delta hepatitis were given different doses of BLV, and HBsAg loss was not achieved in any treatment arm (14). In real-life data, HBsAg loss was not observed in patients receiving BLV (15-18). In conclusion, BLV is safe and well tolerated in the treatment of chronic delta hepatitis, but its effect on reducing HBsAg is limited, and there are no studies with BLV in CHB patients. Therefore, studies are needed for its use in the treatment of CHB.
Targeting cccDNA Directly
CccDNA in the nucleus of infected hepatocytes is the template for HBV transcription and replication, leading to the persistence of HBV infection. Numerous small molecules have been developed as sequence-specific RNA-guided nucleases and proteins that can block the formation of cccDNA while stimulating cell division, enhance its degradation, and silence its transcription (19, 20). These include gene editing using Zn finger nucleases (ZFNs), transcription activator-like (TAL) effector nucleases (TALENs), or clustered regularly spaced short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) system. Designer nucleases can cleave pre-targeted sequences in the HBV genome resulting in predefined mutagenesis, and can inhibit viral gene expression, reducing HBV replication and ultimately the cccDNA pool (4, 20-22).
Another method targeting cccDNA is epigenetic modification. This method renders actively transcribed DNA transcriptionally inactive without changing the nucleotide sequence. It directs DNA-guided epigenetic effectors to predefined sequences of cccDNA for targeted modifications to occur. Potential HBV DNA modifiers include histone acetyltransferases/deacetylases, lysine methyltransferases, protein arginine methyltransferases, and DNA methyltransferases, which act in conjunction with viral factors such as HBx and HBcAg. HBx protein is a good target in this regard (2, 23); HBx hijacks E3 ubiquitin ligase containing cellular DNA damage binding protein 1 (DDB1), causing disruption of the SMC5/6 complex, thereby promoting cccDNA transcription. Pevonedistat, an enzyme inhibitor that activates NEDD8, inhibits the degradation of SMC5/6 in hepatocyte cultures and suppresses viral transcription and protein production. Nitazoxanide, a thiazolide anti-infective agent, has been shown to effectively inhibit the HBx-DDB1 interaction, restore SMC5/6 levels, and suppress viral transcription and protein production in cultured hepatocytes (24, 25).
Apart from these potential HBV DNA modifiers, another method is cccDNA destabilizers (ccc-R08 molecule) targeting the viral genome reservoir. Considering all these cccDNA targeting systems, whether gene editing is reversible or permanent and whether it will affect the host genome or mitochondrial DNA remains to be resolved (26,27).
Targeting Viral Gene Transcription
RNA interference (RNAi) can directly target HBV transcripts and cause degradation. RNAi is a highly specific and efficient method of post-transcriptional gene silencing (27). In this method, synthetic small interference RNA (siRNA) binds with the RNA-induced silencing complex (RISC), and after binding with RISC, targets to cut 10 to 11 bases of specific mRNA small pieces, thereby interrupting the translation process of the specific mRNA and silencing the expression of the target gene. (28).
JNJ-73763989 (JNJ-3989) contains two siRNA triggers targeting HBV RNAs expressed from both cccDNA and HBV DNA integrated into the host genome. Preclinical models resulted in reductions in pgRNA, HBsAg, HbeAg, and HBV DNA in combination with JNJ-3989 monotherapy or ETV. The use of JNJ-3989 alone has shown a good safety profile in healthy volunteers (29). In a phase study, patients who were previously untreated or receiving NA therapy were administered every week (100, 200, or 300 mg subcutaneously), every two weeks (100 mg), or every four weeks (25, 50, 100, 200, 300, or 400 mg). In the last group, in addition to JNJ-3989 treatment every four weeks (200 mg), oral treatment of JNJ-6379 250 mg/day was given for 12 weeks (30). Bersacapavir (JNJ-6379; JNJ-56136379), added to the last group in this study, is an N-class modulator of capsid assembly (CAM-N). JNJ-6379 interferes with capsid assembly, thereby inhibiting pgRNA encapsulation and de novo formation of cccDNA and, ultimately, viral replication (31). When the results of the study were examined, there was no difference in terms of reduction in HBsAg level between the patients who received the dose every 4 weeks and the group who received more frequent doses, a similar decrease in HBsAg was observed at the other dosing except 25 and 50 mg doses, and the patients who took 100-400 mg every 4 weeks. More than 1 log reduction in HBsAg was observed in 97.5%. All patients in the triple combination arm of NA, a siRNA (JNJ-3989), and CAM-N (JNJ-6379) also had a >1 log reduction in HBsAg. The drug was well tolerated, and five non-drug-related serious adverse events were recorded: creatinine elevation, alanine aminotransferase (ALT) and bilirubin elevation. Local side effects at the injection site were other drug-related adverse events, which regressed with treatment discontinuation. In conclusion, in this study, it was shown that the decrease in HBsAg continued in 336 days after the last dose in 336 days of the last dose and was well tolerated by applying JNJ-3989 in combination with an NA, with or without JNJ-6379 every four weeks (30). In another phase 2b study evaluating JNJ-3989, JNJ-6379 (CAM-N, bersacapavir), and nucleotide analog treatment (patients were randomly assigned to receive NA once per day + placebo; oral JNJ-6379 250 mg daily + NA; NA + JNJ-3989 at doses of 40 mg, 100 mg [JNJ-3989 dual 100 mg group], or 200 mg [JNJ-3989 dual 200 mg group], or JNJ-6379 250 mg + JNJ-3989 100 mg + NA) HBsAg seroclearance could not be achieved in any patient; however, JNJ-3989 provided a dose-dependent reduction in HBsAg titer. Nucleotide analog discontinuation criteria were met in 38% of patients in the combination groups containing JNJ-3989 (32).
Another candidate for functional curing siRNA is VIR-2218. This siRNA targets the HBx gene. The functional cure is aimed at combining VIR-2218 with other HBV treatments. VIR-2218 has been shown to inhibit all major transcripts of 10 HBV genotypes. In a phase 2 study, in virologically controlled patients, VIR-2218 was administered subcutaneously at different doses (20, 50, 100, 200 mg in HBeAg positives, 50, 200 mg in HBeAg negatives) under placebo-controlled on the 1st and 29th days, and the patients were treated for 48 weeks followed throughout. While there was a decrease in HBsAg level in the group receiving VIR-2218, there was no change in the placebo arm. At the end of the treatment, HBsAg level decreased 10-fold in 71% (17/24) of the patients, and HBsAg level decreased below 100 IU/mL in 50% (12/24). At the 48th week after the end of the treatment, 23% (4/17) of the patients whose HBsAg level decreased continued to have HBsAg levels below 100 IU/mL, and a sustained response was achieved (all in high-dose patients). The drug was well tolerated at all doses, and no treatment discontinuation because of side effects was observed (33).
Another phase 2 study aimed to increase the HBsAg response with the combination of VIR-2218 with peg-IFN. Virologically suppressed patients were divided into five groups. Peg-IFN α-2a was given in combination with VIR-2218 at different times. The rate of reduction in HBsAg was highest in the group given Peg-IFN α-2a for 44 weeks and VIR-2218 for 13 weeks (-2.9 log10 IU/mL). Again, the highest HBsAg seroconversion was achieved in this group (30% at the end of treatment). No serious adverse events related to VIR-2218 were observed in the patients, and interferon-related side effects were observed in patients receiving Peg-IFN α-2a. As a result of this study, it was stated that the efficacy of VIR-2218 may increase with the combination of immunomodulatory drugs (34). An ongoing phase 2 study of VIR-2218 with VIR-3434 (neutralizing monoclonal antibody against HBsAg), seeking an answer to the question of increasing the efficacy with its combination with immunomodulatory treatments, continues with the combination of VIR-2218, VIR-3434, and Peg-IFN α-2a (35). Another study evaluated the results of the combination of VIR-2218 and ALN-HBV 2 siRNA, and although HBsAg loss was not achieved in any patient, it is hypothesized that VIR-2218, when combined with a targeted immune-stimulating agent, may play a key role in achieving a functional recovery in patients with CHB infection (36).
Another molecule targeting viral transcription is antisense oligonucleotides (ASO). Antisense oligonucleotides are molecular drugs that inhibit gene expression by combining their specific sequences with target gene DNA or mRNA. The molecule in the phase 3 study in this group is bepirovirsen; its mechanism of action is targeting all HBV mRNAs and pregenomic RNAs and causes a decrease in the levels of viral proteins. Specific ASOs bind to complementary HBV RNA transcripts, forming a hybrid ASO/RNA complex that recruits endogenous RNase H, cleaves HBV RNA, and degrades the transcript. HBV causes a decrease in DNA and viral proteins (including HbsAg) (37).
In the phase 2 study, in virologically suppressed and treatment-naïve patients, bepirovirsen was administered subcutaneously to one treatment arm twice a week for the first two weeks, subcutaneously once a week for the next two weeks, and placebo was given to the other group. The patients were followed for the next 26 weeks. In treatment-naïve patients, a decrease in HBsAg level of -1.56 log 10 IU/mL was observed in the group receiving 300 mg bepirovirsen and was found to be significantly higher compared to placebo. A decrease of -1.99 log 10 IU/mL was observed in HBsAg level in the group receiving 300 mg bepirovirsen and in patients receiving NA treatment. HbsAg decreased below detectable limits in four patients who received 300 mg of bepirovirsen, and seroconversion developed in three patients. The most common adverse event in patients receiving bepirovirsen was transient ALT elevations, excluding injection site local side effects, and none of the patients required discontinuation of treatment (37). In another phase 2b study, 10% of 230 treatment-naïve patients who received 300 mg weekly bepirovirsen for 24 weeks, HbsAg clearance developed in 9% of 227 patients who received NA treatment (38). Bepirovirsen is promising for future treatments due to its successful results in clinical trials with high patient numbers, but longer-term studies are needed for efficacy and safety.
Capsid Assembly Inhibitors
The nucleocapsid, which consists of a core protein, is an important structure of HBV. pgRNA and polymerase are first encapsulated before DNA replication takes place. Capsid assembly inhibitors (CpAM) affect viral replication and cccDNA supplementation by blocking nucleocapsid assembly (39). Two classes of CpAM have been defined according to their mechanism of action. Class I CpAMs increase capsid formation kinetics and lead to the formation of misassembled capsids. Class II CpAMs accelerate capsid assembly and form morphologically normal capsids that are empty and lack viral pgRNA and HBV polymerase. Heteroaryldihydropyrimidines (HAPs) type I and phenylpropenamides (PPAs), sulfamoyl benzamides, and some other chemotypes are examples of type II CpAMs (40).
HAP derivatives misdirect core protein dimers to assemble non-capsid polymers, leading to the degradation of the core protein. Morphothiadine (GLS4) is from the HAP family and has a low plasma concentration due to its short half-life. Therefore, the addition of ritonavir (RTV) is aimed at inhibiting hepatic metabolizing enzymes and increasing plasma concentration. The combination of GLS4/RTV was compared with ETV monotherapy for 28 days. Patients were followed up for 40 days. Administration of 120 mg GLS4 for 28 days resulted in reduced serum HBV DNA and HBV pgRNA levels. HBsAg, HBeAg, and HBcrAg reductions were also observed in some patients (41, 42).
ABI-0731 (vebicorvir [VBR] 300 mg orally/day) is an orally administered potent, selective, and pan-genotypic core inhibitor that induces altered, non-functional core protein assembly. A phase 2 study compared the VBR/ETV combination with ETV monotherapy in treatment-naïve HBeAg-positive patients. The reduction in HBV DNA in patients was found to be significantly higher in the combination arm at 12 and 24 weeks. The reduction in pgRNA was significantly higher in the combination arm. No difference was observed between the two treatment arms in terms of serum viral antigens, HBsAg loss, and seroconversion were not observed in any patient, and the combination of VBR and NA was well tolerated (43). In another phase 2 study, VBR/NA combination was compared with NA monotherapy in both HBeAg-positive and HBeAg-negative virologically suppressed patients, and similar results were obtained. Despite the decrease in pgRNA, no decrease was observed in HBsAg compared to NA monotherapy (43, 44).
JNJ-6379 was compared with placebo +NA in treatment-naïve and virologically suppressed patients given 75 mg and 250 mg of NA once daily in combination or as monotherapy for 24 or 48 weeks. The treatment was extended to 48 weeks in patients with a virological response at week 24, and the patients were followed up for 48 weeks at the end of the treatment. Patients who did not meet the elongation criteria were given NA and followed for 24 weeks. Although JNJ-6379+NA provided a significant reduction in 24 HBV DNA and HBV RNA, restricted HBsAg in HBeAg-positive patients resulted in a reduction in HBeAg and did not show superiority in efficacy compared to NA. Extending JNJ-6379 therapy to 48 weeks provides an additional virologically limited contribution. When JNJ-6379 is given as monotherapy, it can lead to the selection of resistant variants. Drug was generally well-tolerated, isolated and transient treatment-emergent ALT and AST increases have been observed during treatment (31, 45). Canocapavir (ZM-H1505R), EDP-514, ABI-H3733, ALG-000184, and RO7049389 are other CpAMs whose phase 1 studies have been completed (46, 47).
HBsAg Release Inhibitors
The most abundant viral antigen in the blood is HBsAg. It is the most important obstacle to the immune control of HBV by the host. Circulating HBsAg is in the form of non-infectious HBV subviral particles. Because these particles are produced independently of viral replication, targeting viral antigens using approved therapies to date has been difficult (2).
Nucleic acid polymer (NAP) REP 2139 inhibits aggregation/secretion of HBV subviral particles. In 12 patients with HDV infection, 15 weeks of REP 2139-Ca, followed by 15 weeks of REP 2139-Ca and Peg-IFN a, followed by 33 weeks of Peg-IFN a treatment, were followed for 3.5 years. HBsAg decreased to <0.005 IU/mL in 5 of 11 patients who completed the study. During the follow-up period, HBsAg continued at <0.005 IU/mL in 3 patients, and anti-HBs seroconversion developed in 4 patients in total (48).
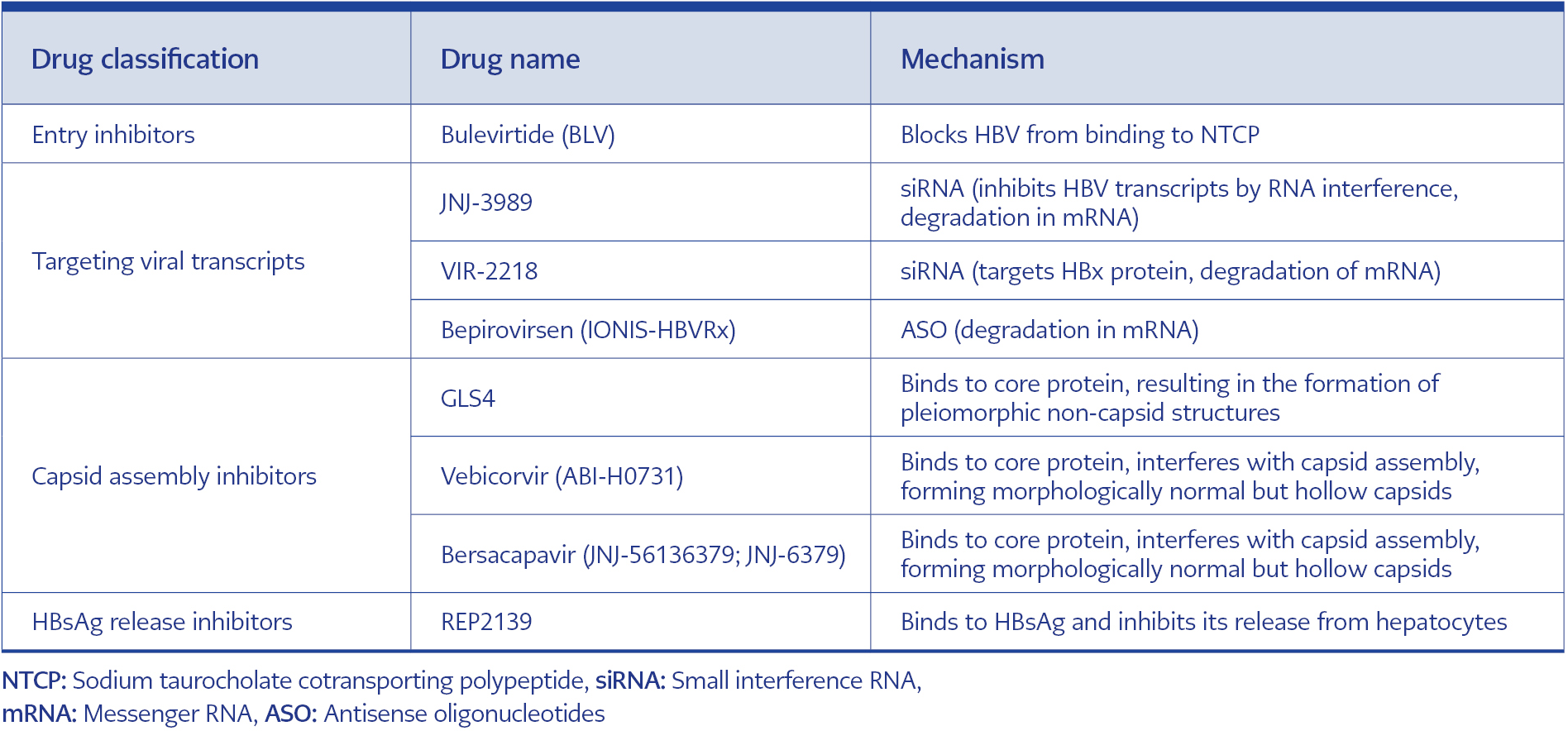
Table 1. New antivirals targeting the HBV life cycle and working mechanism.
Another phase 2 study is the REP 401 study, with either REP 2139-Mg or REP 2165-Mg (250 mg intravenous infusion per week) added to baseline TDF therapy. After 24 weeks of TDF treatment, 40 patients were split into experimental treatment followed by 48 weeks of experimental treatment (TDF + Peg-IFN + REP 2139-Mg or REP 2165-Mg) or 24 weeks of control treatment (TDF + peg-IFN). All patients included in the study were then followed up for a treatment-free 48-week period. In this study, when the group of patients who received TDF + Peg-IFN and those who received NAP in addition to TDF + Peg-IFN were compared at the end of the 48-week period, a decrease in HBsAg level and seroconversion rate were found to be significantly higher in the group with NAP. At the end of the treatment, HBsAg seroconversion was achieved in 60% (24/40) of the patients. At the end of the follow-up period, HBsAg clearance was achieved in 32.5% (13/40) of the patients, and HBsAg seroconversion was observed in 35% (14/40) of the patients. Although more increases in aminotransferases were observed in the group receiving NAP, most of them were associated with a decrease in HbsAg levels and returned to normal at the end of the treatment (49). In a study evaluating the combined treatment of NA, Peg-IFN, and NCT02565719 (NAP, REP 401 study), the functional cure was achieved in 35% of the patients. In this study, the kinetics of HBsAg decrease were also examined, and they showed that a rapid decrease in HBsAg was achieved, especially with Peg-IFN treatment, and the chance of functional cure increased in the group in which the rapid decrease was achieved (50). DAA, whose studies are ongoing in the treatment of HBV, is summarized in Table 1.
Immunomodulatory Therapies
Checkpoint Inhibitors
The liver is an organ with very important functions in the immune system, and immune activities in the liver play a critical role in virus clearance. However, intrahepatic immunity is prone to immunotolerance during chronic HBV infection. Programmed cell death protein-1 (PD-1) and PD-ligand 1 (PD-L1) act as inhibitory co-stimulatory molecule, leading to inhibition of the immune response. Studies have shown improvement in HBV-specific T-cell functions by inhibiting the PD-1/PD-L1 interaction in vitro. It was observed that CD8+ T-cell proliferation was also increased by blocking the PD-1/PD-L1 interaction. Studies provide a new approach in which T-cell functions can be improved in patients with chronic HBV infection, with new immunotherapeutic approaches to be developed. As a result, the use of agents effective in this step can restore T-cells. Molecules with anti-PD-1/PD-L1 activity for chronic HBV infection may be promising therapeutic candidates that contribute to improving T-cell functions not only in peripheral but also in intrahepatic lymphocytes (51, 52). Nivolumab, programmed cell death protein-1 (PD-1) is a molecule developed as an immune checkpoint inhibitor. Combining lenvatinib with a PD-1 inhibitor has been explored for the treatment of unresectable hepatocellular carcinoma (uHCC). In a study, lenvatinib + PD-1 inhibitor (nivolumab, pembrolizumab, camrelizumab, etc) treatment demonstrated long survival in 378 uHCC patients (HBV was the etiological cause in 89.9% of uHCC) (53).
In another phase 1 pilot study conducted recently, in which HBeAg-negative CHB patients were evaluated. The safety and immunological efficacy of nivolumab therapy given alone and in combination with a therapeutic HBV vaccine, GS-4774, were evaluated. At 24 weeks, 14% (3/22) of the patients had a >0.5 log10 decrease in HBsAg levels, and HBsAg loss was detected in one patient using the combination, and the treatments were well tolerated. Thus, these pilot studies support the inclusion of PD-1/PD-L1 blockade in future combination strategies for functional cure therapy of chronic HBV infection (4, 54).
Monoclonal Antibodies
Various monoclonal antibodies targeting HBV have been developed in recent years, showing high affinity, specificity, and neutralizing effects. Further studies confirming immunotherapy for HBV infection may require supplementation with broadly neutralizing antibodies. Activated B cells further limit the spread of HBV infection by producing neutralizing antibodies, preventing viral spread, and clearing circulating viruses. In conclusion, monoclonal antibodies may be a promising alternative to preventing and treating HBV infection (4, 55, 56).
GC1102 and VIR-3434 are monoclonal antibodies. The results of the studies show that combining GC1102 with antiviral drugs can reduce HBsAg titers and increase the chances of functional cure in patients with CHB. In addition, it was reported that 3000 mg single dose VIR-3434 treatment was well tolerated in healthy participants (5759). Preliminary data from the Phase 1b study showed that even a single low dose (6 mg) of VIR-3434 in CHB patients produced rapid HBsAg reduction and was maintained for two weeks after administration. In addition, no safety issues or serious adverse events were observed in healthy volunteers at doses up to 3000 mg. This significant reduction in HBsAg suggested that VIR-3434 has the potential to play an important role in the functional treatment of chronic HBV infection (4, 59).
Engineered T-Cells and Other Immunomodulators
The efficacy of immunotherapeutic approaches to CHB patients has been limited, probably because HBV-specific T-cells are rare and have a depleted phenotype. Studies have shown that mitochondrial defects limit the metabolic functions of HBV-specific depleted T-cells. In this respect, additional cytokines may have some effect. Recent studies have aimed to restore the immune system through adaptive transfer of specially designed HBV-specific T-cells. Therefore, mitochondria-focused strategies represent promising targets for the treatment of chronic HBV infection (4, 60-62).
Experimental studies with a precursor molecule, IMC-I109V, supported its use in clinical studies. IMC-I109V, monoclonal T-cell receptors that upregulate the immune system against the virus, represents a therapeutic strategy combining an affinity-enhanced T-cell receptor with an anti-CD3 T-cell activating moiety. The data obtained show that these agents become a promising therapeutic option for the treatment of CHB patients (4, 63). In the phase 1/2 study, HBsAg declined in virologically suppressed patients who underwent IMC-I109V. Administration of a single dose of IMC-I109V was not associated with cytokine release syndrome or any other adverse event (64).
Toll-Like Receptor Agonists
Toll-like receptors (TLRs), which are the first line of defense against pathogens, are functionally involved in the recognition of antigens against and against them, maturation of dendritic cells, and initiation of antigen-specific adaptive immune responses (65, 66). By binding interferons (IFNs) and other cytokines/chemokines to TLRs, natural killer (NK) cells and cytotoxic T lymphocytes are stimulated, thereby activating innate and adaptive immune responses simultaneously (66). TRL7 and TRL8 agonists are involved in the production of endogenous IFNs, induction of IFN-stimulated genes, and activation of other signaling cascades such as the Janus kinase/signal converter and activator of the transcription signaling pathway. It leads to inhibition of hepatitis B virus replication (67).
Vesatolimod (GS-9620), a TRL7 agonist, increases T-cell and NK-cell responses and decreases the ability of NK to suppress T-cells (68). In a phase 2 study, it was reported that GS-9620 was safe and well tolerated in patients with chronic hepatitis B, but no significant decrease in HBsAg was observed (69). In a meta-analysis of 391 studies, GS-9620 in the treatment of HBV was associated with only mild side effects; It was found to be safe and tolerable (70). Other TLR7 agonists with clinical trials, such as RO7020531 and TQA3334 (JNJ-4964), have demonstrated the possibility of suppressing HBV-specific immunity (71, 72).
Selgantolimod (GS-9688), a TLR8 agonist, has the potential to increase responses, contributing to viral control and modulation of regulatory mediators (73). In a randomized, double-blind, placebo-controlled phase 1b study, GS-9688 was found to be safe and well tolerated in CHB patients and elicited cytokine responses (74). In a phase 2 clinical trial, 24 HBeAg-positive and 24 HBeAg negative CHB patients were evaluated after 24 weeks of GS-9688 treatment with a NA followed by 24 weeks of NA monotherapy. In the American Association for the Study of Liver Disease (AASLD) 2020 study, HBsAg loss was reported in 2 patients and HBeAg loss in 3 patients at week 48 (75). In another phase 2 clinical study, 67 HBeAg-positive/negative CHB patients were randomized to receive TAF with GS-9688 3 mg, 1.5 mg, and placebo once weekly for 24 weeks (2:2:1). In the European Association for the Study of the Liver (EASL) 2021 study, it was reported that ≥0.5 log10 IU/mL HBsAg reduction was achieved in three patients at week 24 and in four patients at week 48 in the study under GS-9688 treatment (76).
Therapeutic Vaccines
Immunogenic vaccines and T-cell peptide vaccines, when used alone or with oral antiviral therapy, have previously demonstrated sustained and protective HBV-specific B- and T-cell responses in HBV-naïve patients and reduced hepatitis B virus replication in animals with chronic hepadnaviral infection; however, has no efficacy in patients with CHB (77-79). Therapeutic vaccines aimed at stimulating the patient’s immune system to combat infectious pathogens show promise as an immunotherapeutic approach, but many therapeutic vaccines developed have failed to provide HBV-specific immunity in most patients (80-82). Adverse outcomes from therapeutic vaccines are most likely due to stimulation of antibodies rather than stimulation of cytotoxic T-cell responses (80). Therapeutic vaccines have been used together with existing antiviral drugs in most clinical trials (4).
GS-4774 is a recombinant, heat-killed Saccharomyces cerevisiae yeast-based vaccine that expresses HBV-specific antigens, including HBsAg, HbcAg, and hepatitis B virus X protein. In a phase 2 study, GS-4774, in combination with TDF, was evaluated for its ability to enhance the anti-HBV immune response. Researchers have found that GS-4774 can increase the production of IFNγ, tumor necrosis factor (TNF), and interleukin 2 by CD8+ T-cells exposed to antigenic peptides. Although GS-4774 does not reduce HBsAg levels in patients, its strong immunostimulatory effect on CD8+ T-cells can be combined with other antiviral agents to increase the antiviral immune response (83).
HeberNasvac (ABX-203) is a vaccine containing both HBsAg and HBcAg formulated for intranasal administration. The results of a phase 3 clinical trial of ABX-203 were disappointing, with no significant superiority in reducing HBV DNA and normalizing liver function (84) Another phase I clinical trial in Cuba involved six CHB patients who were unresponsive or incompletely responding to IFNα, and HBeAg loss was demonstrated in three HBeAg-positive patients after five years of follow-up. Undetectable viral load was obtained in five patients, and liver stiffness measurements were found to be <7.8 kPa in all of them (85). This vaccine is currently being modified, but more studies on efficacy and safety are needed (86).
In preclinical and early clinical studies, BRII-179 was characterized by the induction of a Th1-type immune response (87). In the EASL 2021 study, in a phase 1b/2a clinical study for the treatment of CHB patients, it was reported that the therapeutic candidate BRII-179, alone or in combination with IFNα, was able to induce B and T-cell immune responses simultaneously and gave good results. A clinical phase 2 study of BRII-179 in combination with BRII-835 siRNA (VIR-2218) is ongoing HBV (88). Preliminary data indicate that therapeutic vaccines will be effective when administered with a combination approach. Preclinical studies on other therapeutic vaccine candidates are ongoing (89, 90).
Apoptotic Protein Inhibitor Antagonists
Members of the apoptotic protein inhibitors (APIs) family were first thought to be functionally limited by inhibition of apoptosis. With further research, it became clear that APIs are not only custodians of cell death but may also play a role in the regulation of inflammation and innate and acquired immunity. Results from animal studies revealed that APIs play an important role in T-cell proliferation and survival in the inflammatory environment of viral infection, suggesting that API antagonists may interfere with immune responses (91). API antagonists were initially developed to promote the apoptosis of cancer cells. Death ligands such as TNF can dichotomously promote cellular activation and survival or apoptosis. API antagonists constrain the ability of TNF to engage the apoptotic pathway and instead promote the activity of inflammatory pathways (92). HBV causes upregulation of TNFR1 protein expression in infected cells and promotes local TNF production. Therefore, API antagonists may be an option on the road to cure in HBV treatment (93).
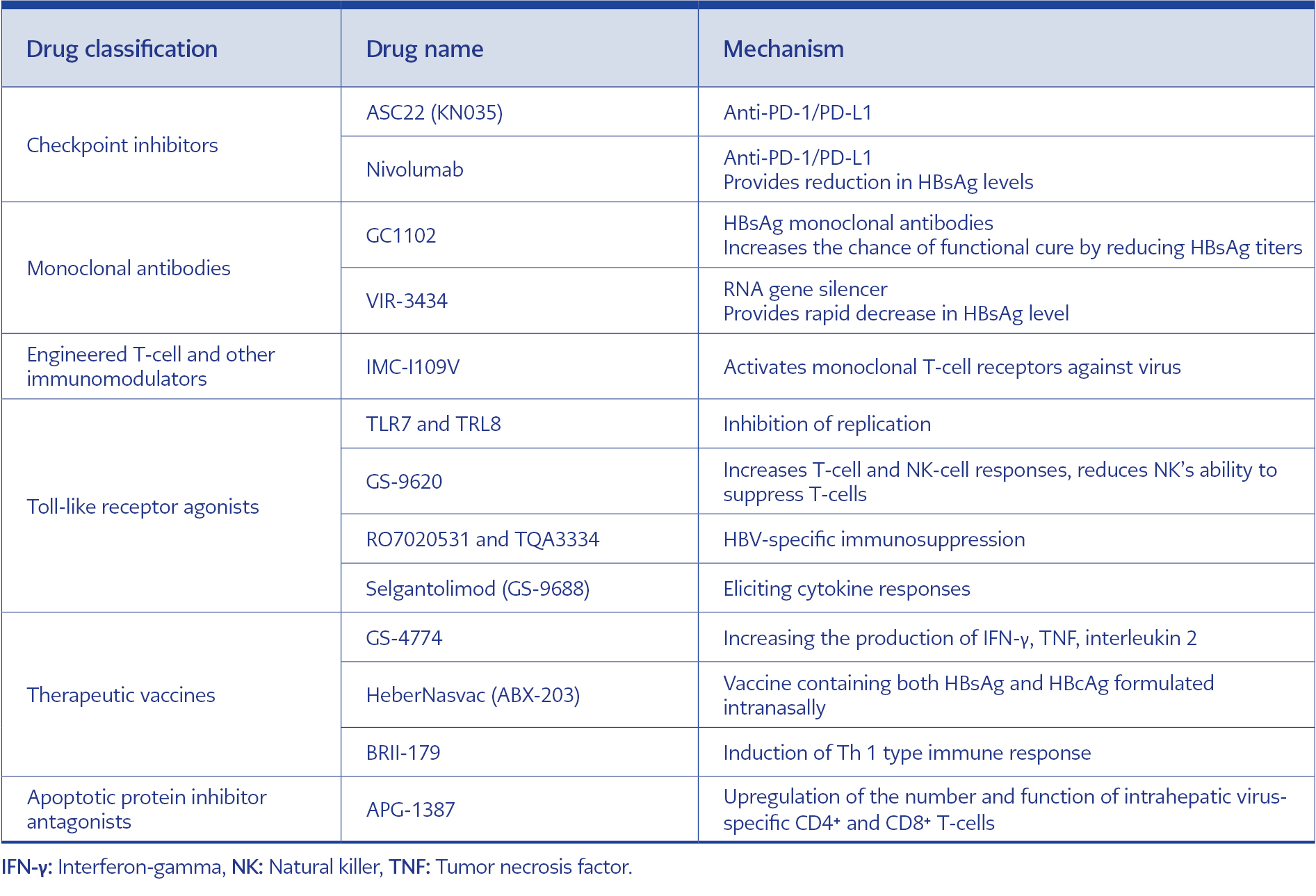
Table 2. Immunomodulatory treatment options and working mechanisms.
APG-1387 is API antagonist-based new drug for the treatment of hepatitis B, and its mechanism of action includes downregulating APIs by mimicking the endogenous second mitochondria-derived activator of caspase molecules, thereby triggering and accelerating the apoptosis process. Preclinical findings demonstrated that APG-1387 could clear chronic HBV infection in various mouse models through a unique mechanism of apoptosis induction and immune regulation. Further investigation revealed that this clearance mechanism may be associated with upregulation of intrahepatic virus-specific number and function (94). Immunomodulatory treatment options and working mechanisms are given in Table 2.
Conclusion and Future Perspectives
The current approach in the treatment of chronic HBV infection is to use more than one drug combination. Decreased HBsAg load with antiviral treatments targeting the viral life cycle also provides an opportunity for immune restructuring and can help to eliminate T-cell depletion. As new DAA and immunomodulatory therapies continue to emerge and evolve, a functional cure in HBV treatment may be an achievable goal.