Introduction
The increase of colistin resistance in Acinetobacter baumannii is a great concern in various regions of the world such as Asia, Europe, and North and South America (1). Exposure to colistin is considered the most significant factor for emerging of colistin resistance (2); however, details of the in vivo response of A. baumannii to colistin exposure, such as the number of days for the development of resistance was not described. The molecular studies about colistin resistance indicated two main mechanisms: modifications in lipid A structure and complete loss of LPS (3). In this study, we analyzed the adaptation of A. baumannii to colistin use on a disease course of a patient. The cellular effects of colistin use were mimicked in laboratory conditions to understand the relationship between the clinical features and molecular resistance mechanisms.
Method
Case History
A 45-year-old woman had a stab wound caused by a sharp object penetration of the abdominal and thoracic walls. Two days after abdominal and thoracic surgery in another hospital, she was transferred to our intensive care unit (ICU). On the third day of hospitalization, her body temperature was elevated, and her white blood cell count was 27,280 /µL, C-reactive protein was 356.9 mg/L and procalcitonin was 0.86 ng/mL. Carbapenem-resistant A. baumannii (CarR-AB) was isolated from the cultures of sputum and abdominal drainage on the fifth day. Colistin 300 mg/day and meropenem three g/day were started, but clinical and laboratory responses were poor. On the 15th day of hospital stay, CarR-AB was isolated again from abdominal drainage. Meropenem was switched to tigecycline 100 mg/day, and colistin was maintained. On the 29th day, antibiotic treatment was stopped based on clinical and laboratory findings. On the 38th day, colistin-resistant A. baumannii was isolated from the abdominal drainage. A. baumannii was only susceptible to tigecycline and amikacin. Colistin 300 mg/day and tigecycline 100 mg/day were started, however, colistin was switched to amikacin in the 48th day because of the generalized itching that does not respond to anti-histaminic treatment. During the therapy with amikacin and tigecycline, she had generalized tonic-clonic seizures probably associated with amikacin on the 50th day. She had no fever, her white blood cell count was 12,420 /µL, C-reactive protein was 30.4 mg/L and procalcitonin was 0.24 ng/mL on the seizure day, and antimicrobial treatment was stopped. In the following days, she had no fever, and she was discharged on the 72nd day of hospitalization. At year 1 of follow-up, the patient was completely free of symptoms. At year 2 of follow-up, she was completely well, and follow-up laboratory tests were within normal limits.
Microbiological Studies
Four colistin susceptible A. baumannii from sputum (K399, K411) and intra-abdominal fluid (IAF) (K408, K412) were isolated. After 25 days of colistin use, a colistin-resistant A. baumannii (K409) was isolated from IAF. The clonal relatedness of the isolates was determined by the repetitive PCR (rep-PCR) based
Diversilab system (Biomerux, France). The colistin minimum inhibitory concentration (MIC) of each isolate was determined by broth microdilution following the Clinical Laboratory Standards Institute guidelines (4).
The four colistin susceptible isolates were sub-cultured daily on Mueller Hinton agar (MHA) (Becton, Dickinson and Company, U.S.) containing 1 µg/mL colistin until the 40th generation. All generations were tested for colistin MIC’s and alterations in pmrCAB and Lpx genes. Mutations in lpxA, lpxC, lpxD, and pmrCAB were detected by Sanger Sequencing (Applied Biosystems ABI 3500) using previously described primers (5, 6). ABI files were analyzed in Applied Maths Bionumerics version 7.5 Software (Biomerieux, France). The expressions of pmrCAB were studied by qRT PCR (LightCycler 480 II, Roche, Germany) using previously described primers (5, 6). The housekeeping gene was 16SrRNA, and A. baumannii ATCC 19606 was used to normalize Ct values. Carbapenemase typing and mcr-1 positivity were studied by PCR.
Results
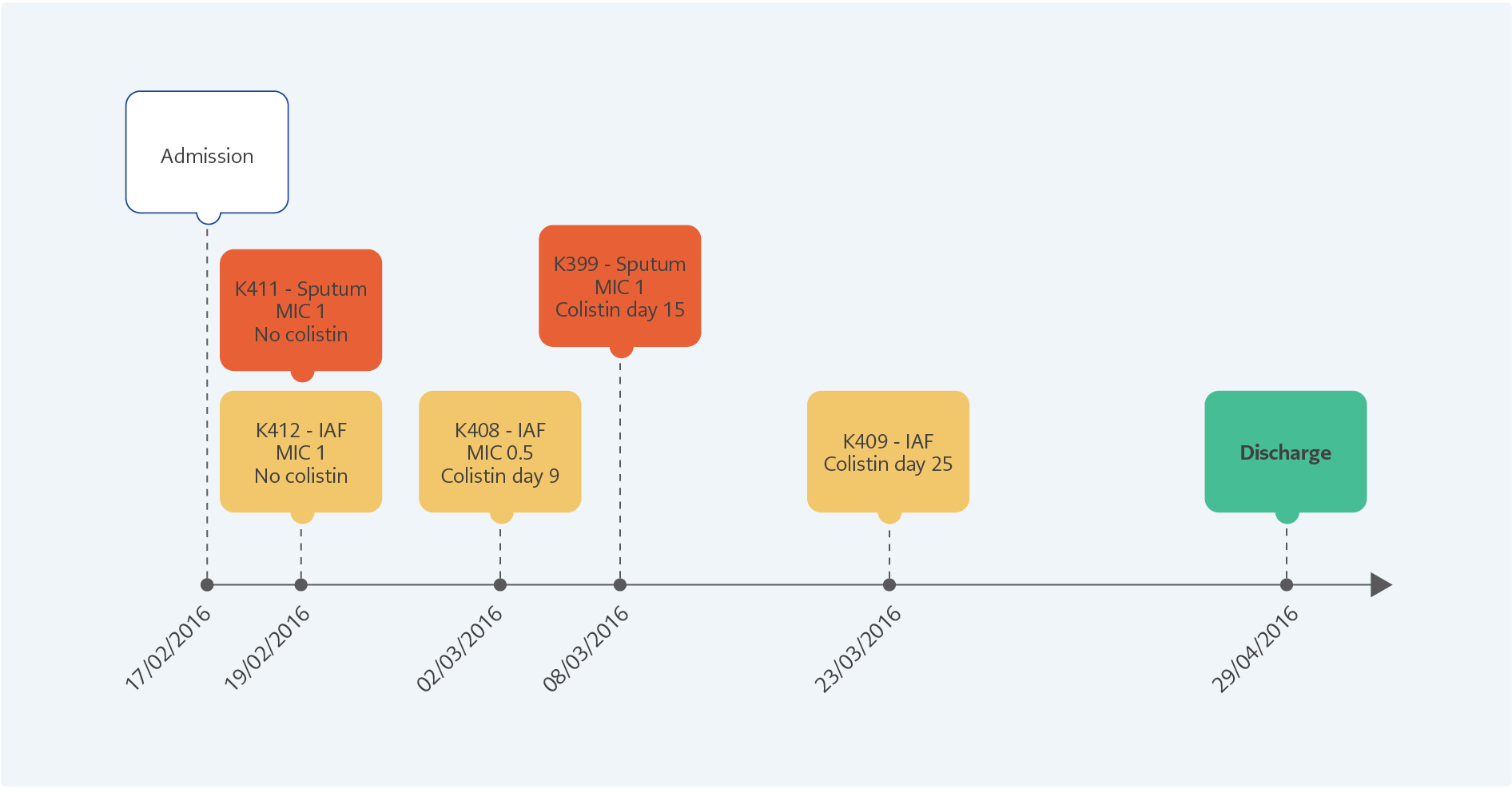
Figure 1. The clinical progression of the patient from admission to discharge of the hospital. The K411 and K412 were isolated before colistin treatment, K408 and K399 were at the 9th and 15th days of the therapy. The colistin-resistant K409 was isolated five days after stopping 25 days of colistin therapy.
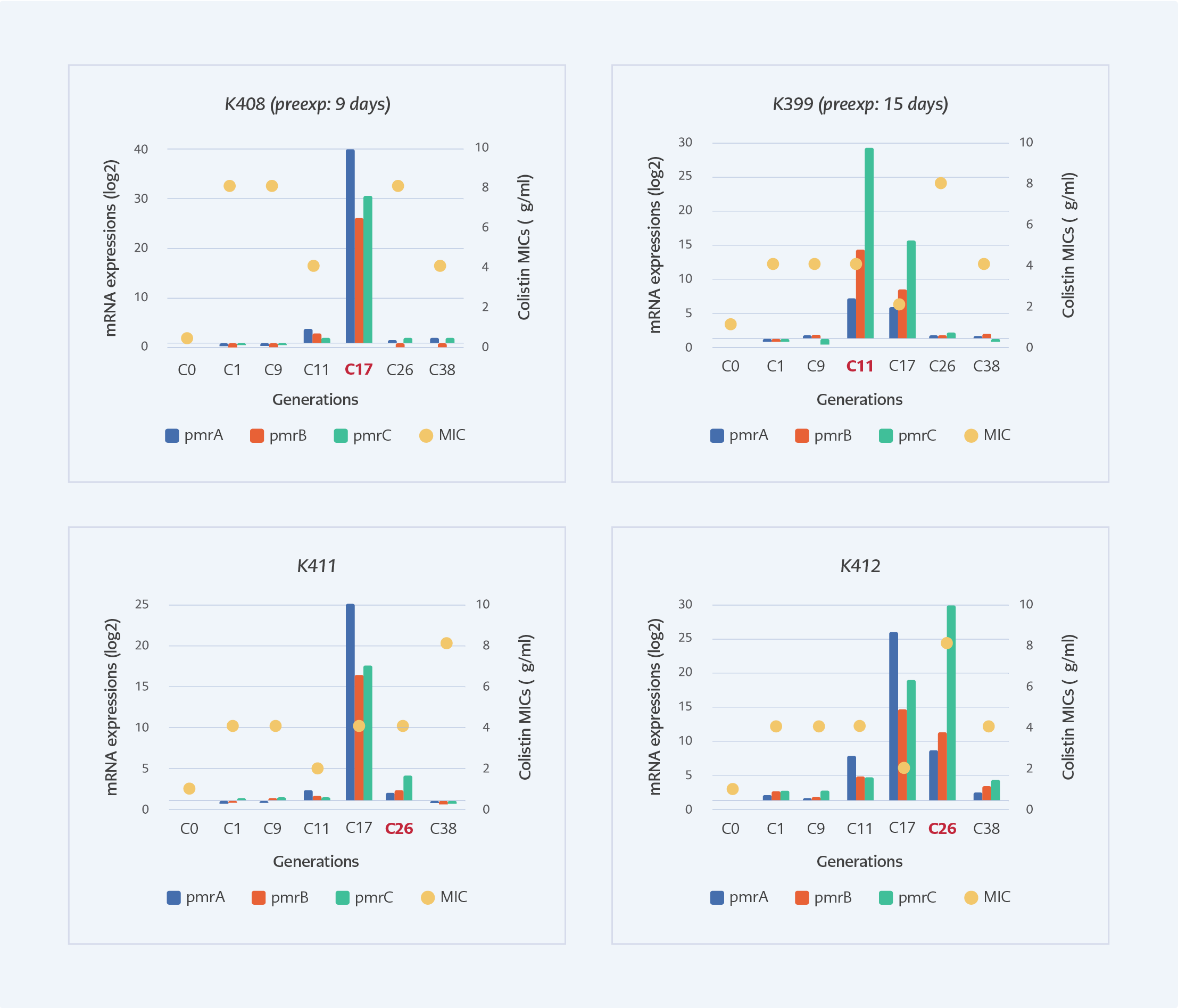
Figure 2. The MIC levels and pmrCAB expressions of selected subculture generations of susceptible isolates under colistin exposure. Red texts indicate the generation that corresponds to the clinical colistin exposure time.
The clinical progression of the patient is shown in Figure 1. After initiating the colistin therapy, two susceptible isolates (K399 and K408) were detected on the 9th and 15th days. The colistin-resistant A. baumannii (K409) was isolated after 25 days of therapy. All the isolates were clonally related with >95% similarity and positive for blaOXA-23. No mcr-1 was detected. In the colistin-resistant isolate (K409), multiple insertions were found in different regions of pmrA, pmrB, pmrC, lpxA, lpxC, and lpxD genes. The pmrA, pmrB, and pmrC genes were also found to be 1.6, 1.74 and 1.72-fold overexpressed compared to the susceptible K408, respectively.
In the laboratory, the pmrCAB expressions of susceptible isolates were significantly induced at the generations that correspond to the 26th day of colistin exposure which was the 11th generation for K399, the 17th generation for K408, and the 26th day for K412. Colistin MIC’s increased above 2 mg/L after the first generation and the highest MIC of all generations was 8 mg/L (Figure 2).
Discussion
Colistin is one of the last resorts treating of MDR A. baumannii infections; however, resistance against colistin is increasing rapidly (7). Many researchers reported the development of colistin resistance either in a laboratory environment or in clinical settings; however, the causes of resistance are still not well known. This study focuses on colistin resistance mechanisms of A. baumannii induced by in-vivo and in-vitro colistin exposure.
In our study, after 25 days of colistin treatment, colistin-resistant A. baumannii was isolated from the intra-abdominal fluid of the patient. Colistin exposure is found to be the main risk factor for the development of colistin resistance by previous studies (2, 8). We mimicked the effects of colistin exposure in the laboratory by measuring MIC and expressions of pmrCAB. After the first passage in the laboratory, colistin MIC increased above the resistance breakpoint (>2 mg/L). (Figure 2). A recent study also reported phenotypically colistin-resistant mutants of A. baumannii obtained after 24 hours of colistin exposure (9). However, after the 25th day of colistin therapy, we isolated colistin-resistant A. baumannii from the patient. Molecular analyses of generations in the laboratory supported this finding, and the expressions of pmrCAB in generations reached the highest level at the 11th generation of K399, 17th generation of K408 and 26th generation of K411, which were closely corresponded to the duration of therapy (Figure 2). The development of colistin resistance is an adaptation to environmental stress (10); therefore, it needs a period for the regulation of genetic and metabolic activities. It was shown that mutations and regulation of expression patterns are gained by stress-exposed bacteria in a certain period (11). Lee and colleagues investigated induced colistin resistance in P. aeruginosa and found that six days of 4 mg/L of colistin exposure was sufficient to gain colistin-resistant phenotype in vitro (12). The expressions of pmrA, pmrB and pmrC genes of colistin-resistant (K409) isolate were also found to be 1.6, 1.74 and 1.72-fold higher compared to susceptible one, respectively. Beceiro et al. reported increased expression of pmrA (4- to 13-fold), pmrB (2- to 7-fold) and pmrC (1- to 3-fold) in resistant A. baumannii (6).
We conclude that the duration of colistin exposure is critical for the adaptation and establishment of colistin-resistant phenotype. A. baumannii can develop a stable colistin-resistant phenotype with elevated MICs and upregulated pmrCAB operon after three weeks of colistin exposure.